
Revista
Científica UDO Agrícola Volumen 10. Número 1. Año 2010. Páginas: 103-108. Nota
Técnica
Extracción
y cuantificación de ADN de pajillas de semen bovino criopreservado
Extraction and
quantification of DNA bovine of straws semen criopreserved
Benigno
RUÍZ SESMA ![]() ,
Reyna Isabel ROJAS MARTÍNEZ, Horacio RUÍZ HERNÁNDEZ, Paula MENDOZA NAZAR, María
Angela OLIVA LLAVEN, Carlos Enrique IBARRA MARTÍNEZ,
Gabriela AGUILAR TIPACAMU, José Guadalupe HERRERA HARO, Alfonso HERNÁNDEZ
GARAY, Diana SANZON GÓMEZ, Gerardo Uriel BAUTISTA TRUJILLO, Alfonso de Jesús
RUÍZ MORENO y Leopoldo M. MEDINA SANZON
,
Reyna Isabel ROJAS MARTÍNEZ, Horacio RUÍZ HERNÁNDEZ, Paula MENDOZA NAZAR, María
Angela OLIVA LLAVEN, Carlos Enrique IBARRA MARTÍNEZ,
Gabriela AGUILAR TIPACAMU, José Guadalupe HERRERA HARO, Alfonso HERNÁNDEZ
GARAY, Diana SANZON GÓMEZ, Gerardo Uriel BAUTISTA TRUJILLO, Alfonso de Jesús
RUÍZ MORENO y Leopoldo M. MEDINA SANZON
Facultad Medicina Veterinaria y Zootecnia, Universidad
Autónoma de Chiapas. Rancho San Francisco Km 8 Carretera Ejido Emiliano Zapata,
Tuxtla Gutiérrez, Chiapas. México.
E-mails: brsesma@prodigy.net.mx y brsesma@colpos.mx ![]() Autor para correspondencia
Autor para correspondencia
|
Recibido: 30/08/2009 |
Fin de arbitraje: 15/09/2009 |
Revisión recibida: 11/01/2010 |
Aceptado: 20/01/2010 |
RESUMEN
El semen criopreservado
viene diluido con Tris, ácido cítrico, fructosa, glicerol, leche descremada en
polvo y yema de huevo. Para extraer el ADN del semen es necesario eliminar el
ADN exógeno, esto no se logra con las técnicas normales de extracción con semen
fresco, por lo que es necesario recurrir a las técnicas forenses. El objetivo
del presente estudio fue evaluar tres protocolos para la extracción de ADN de
semen bovino criopreservado. El estudio se realizó en
el laboratorio de fitopatología vegetal del Colegio de Postgraduados. Se
utilizaron pajillas de ½ mL y ¼ mL.
Las variables evaluadas fueron; Tiempo de ejecución del protocolo (T), pureza
del ADN y concentración de ADN. Las técnicas evaluadas fueron: Tratamiento1: Yoshida et al.
(1995). Tratamiento2: Penacino (1997). Tratamiento3:
Técnica rápida de Penacino (1997). El menor tiempo
para ejecución del protocolo fue para el tratamiento tres con 100 minutos. Los
carriles 1 al 4 y 7 al 10, se observa la presencia de proteína. Cuando se
utilizaron una o dos pajillas de ¼ mL se observa poca
proteína y mejor calidad del ADN. En carriles del uno al 10, los valores de la
relación A260/A280 son inferiores 0.1. Los carriles 12 y
17 presentan valores cercanos al rango de pureza. Se concluye que para la
extracción de ADN de semen bovino criopreservado se
debe utilizar una pajilla de ¼ mL por muestra con la
técnica Penacino (1997), y dos pajillas de ¼ mL por muestra para la técnica rápida Penacino
(1997).
Palabras clave: Semen, toros, criopreservado, extracción,
ADN
ABSTRACT
The semen criopreserved
comes diluted with tris, citric acid, fructose, glicerol, milk skimmed in powder and yolk of egg. To
extract the DNA of the semen it is necessary to eliminate the ADN exogen, this is not achieved with the normal technique of
extraction with fresh semen, for what it is necessary to resort to the forensic
techniques. The objective of the present study was to evaluate three protocols
for DNA's extraction of bovine semen criopreserved.
The study was realized in the laboratory of vegetable fitopatology
of Colegio de Postgraduados.
They were in use straws of ½ mL and ¼ mL. The
evaluated variables were; Time of execution of the protocol (T), purity of the
DNA and DNA's concentration. The evaluated techniques were: Treatment1: Yoshida
et al. (1995). Treatment2: Penacino (1997). Treatment3: Penacino
(1997)'s rapid Technique. The minor time for execution of the protocol was for
the treatment three with 100 minutes. The rails 1 to 4 and 7 to 10, is observed
the presence of protein. When one or two was in use straws of ¼ mL is observed
few protein and better quality of the DNA. In rails of one to 10, the values of
the relation A260/A280 are low 0.1. The rails 12 and 17 present values near to
the range of purity. There concludes that for DNA's extraction of bovine semen criopreserved must use a straw of ¼ mL for sample with the
technique Penacino (1997), and two straws of ¼ mL for
sample for the rapid technique Penacino (1997).
Key words: Semen, bulls, criopreserved,
extraction, DNA
INTRODUCCION
La utilización de semen bovino criopreservado en pajillas permite la diseminación de genes
de reproductores con un mérito genético superior en los sistemas de producción,
sin embargo, la mayoría de estos toros carecen de estudio en la cual se
identifique los genes de importancia económica como el gen leptina,
caseína, etc. Para identificar los genes que transmiten los toros, es necesario
la extracción del ADN, sin embargo, muchas veces no se tiene acceso a los toros
y la única fuente para la obtención del ADN es el semen criopreservado,
este generalmente viene diluido con Tris, acido
cítrico, fructosa, glicerol, leche descremada en polvo y yema de huevo
(Olivares y Urdaneta, 1985; Boeta y Zarco 2000;
Hernández et al. 2003; Medina-Robles et al. 2007), esto ocasiona que en la
pajilla de semen se tenga ADN exógeno y el ADN del espermatozoide de toros, por
lo que es necesario eliminar el ADN exógeno antes de extraer el ADN del
espermatozoide, esto no se logra con las técnicas que normalmente se utilizan
para la extracción de ADN de semen
fresco, por lo que es necesario recurrir a las técnicas forenses. Por lo tanto,
el objetivo del presente estudio fue evaluar tres protocolos para la extracción
de ADN de pajillas de semen bovino criopreservado.
MATERIALES Y MÉTODOS
La evaluación de los tres protocolos de
extracción y purificación de ADN a partir de pajillas de semen bovino criopreservado se realizó en el Laboratorio de Fisiología y
Biología Molecular del Colegio de Postgraduados, Campus-Montecillo. Se
utilizaron pajillas de semen bovino criopreservado de
0.5 mL y 0.25 mL.
Las variables evaluadas fueron; Tiempo de
ejecución del protocolo (T), pureza del ADN en gel de agarosa (Pur) y concentración de ADN. Los tratamientos evaluados
fueron los siguientes:
Tratamiento
1
Técnica descrita por Yoshida
et al. (1995) que
consiste en: Paso 1; Se colocaron 2 pajillas de semen bovino criopreservado de 0.5mL o 0.25 mL
en un tubo eppendorf de 1.5 mL,
agregar 0.5 mL de solución de lisis 1 (Bufer TNE : 10 mM Tris-HCl (pH 8.0), 10 mM tetraacetato etilenediamina
(EDTA), 100 mM cloruro de sodio con 1% sulfato dodecil
de sodio (SDS) y 100 µg./ml Proteinasa K). Los tubos con la solución de lisis 1 se
encubo en baño maría por 3 horas a 70oC. Después de la incubación
los tubos se centrifugaron a 14000 rpm por 10 minutos, se tiró el sobrenadante
que contenía ADN del huevo de gallina y otras impurezas. Las cabezas del los espermatozoides quedaron en la pastilla en el fondo
del tubo, a este se le agrego 0.1 ml de búfer de lisis TNE y se incubo en baño
maría por 1 hora a 70oC para
asegurar la degradación de ADN extraño. Paso 2: En este paso se rompen las
cabezas de los espermatozoides para liberar el ADN. Una vez retirado los tubos
del baño maría, se le agrega 0.5 mL de solución de búfer de lisis TNE con 1% sulfato dodecil de sodio (SDS), 100 µg./ml Proteinasa K y 0.04 M Dithiotreitol (DTT), se incuba en baño maría por 8 horas a
56oC. Esta segunda digestión contiene el ADN de los espermatozoides
principalmente. A cada tubo se le añadió 750 µl fenol, se mezcló en vortex (Modelo MS1 Minishaker, Marca
IKA®) durante 30 segundos, se centrifugo (Modelo Spectrafuge
16M, Marca Labnet®) a 14000 rpm por 5 min. Se extrajo
750 µl del sobrenadante y le le agrego 750 µl
de fenol, se mezcló en vortex durante 30 segundos, se
centrifugo a 14000 rpm por 5 min. Nuevamente se extrajo 750 µl del sobrenadante y le aplico 750 µl de cloroformo/alcohol isopropilico (24:1), se mezcló en vortex
durante 30 segundos, se centrifugo a 14000 rpm por 5 min. Del sobrenadante se
extrajo 600 µl teniendo el cuidado de
no extraer impurezas, a esto se le aplico
600 µl cloroformo/alcohol isopropilico (24:1), se mezcló en vortex
durante 30 segundos, se centrifugo a 14000 rpm por 5 min. Se extrajeron 350 µl de sobrenadante y le agrego 35 µl (10% del volumen) de acetato de sodio
(pH 5.3) y se le adiciono 875 µl de
etanol puro (2.5 veces el volumen), se mezcló en vortex
durante 30 segundos, se centrifugo a 14000 rpm por 15 min. El sobrenadante se
decantó y la pastilla se dejó secar en la incubadora (Boekel
Scientific; BOEKEL®) a 37oC por 1 hora,
finalmente, se resuspendió en 30 µl de agua destilada
estéril y se guardaron a –4oC para su medición posterior.
Tratamiento
2
Técnica descrita por Penacino
(1997). Paso 1: Se colocaron 2 pajillas de semen bovino criopreservado
de 0.5 mL o 0.25 mL en un
tubo eppendorf de 2 mL,
para la separación del material utilizado como diluyente (yema de huevo) a cada
tubo se le agrego 40 µl Proteinasa
K (Se disolvió 10 mg de proteinasa K liofilizada en 2
ml de agua destilada estéril), 200 µl
de sulfato dodecil de sodio (SDS) 10 % (se disolvió
50 gr de dodecil sulfato de sodio en 450 ml de agua
caliente, se ajustó el a pH 7.2 por agregado de unas gotas de ácido clorhídrico
concentrado y se aforo 500 ml) y 1000 µl
de TEC (se mezcló 100 ml de [1 ml de TRIS/ClH 1M pH:
7.5 con 2 ml de EDTA disódico 0.5M pH: 8, aforado a
100 ml con agua destilada] con 3.3 ml de cloruro de sodio 3M). Los tubos se
mezclaron en vortex durante 30 segundos y se incubo
en baño maría durante 2 horas a 56 ºC. Se centrifugo
a 14000 por 10 minutos y se retiró el sobrenadante, que contiene ADN extraño.
Paso 2: El precipitado del paso anterior (espermatozoides), se resuspendió nuevamente en 40 µl Proteinasa K (Se disolvió 10 mg de proteinasa K liofilizada en 2 ml de agua destilada
estéril), 200 µl de sulfato dodecil de sodio (SDS) 10 % (se disolvió 50 gr de dodecil sulfato de sodio en 450 ml de agua caliente, se ajustó
el a pH 7.2 por agregado de unas gotas de ácido clorhídrico concentrado y se
aforo 500 ml) y 1000 µl de TEC (se mezcló
100 ml de [1 ml de TRIS/ClH 1M pH: 7.5 con 2 ml de
EDTA disódico 0.5M pH: 8, aforado a 100 ml con agua
destilada] con 3.3 ml de cloruro de sodio 3M y se agregó 10 µl de Dithiotreitol
DTT (se disolvio 0.154 gr. de ditiotreitol
en 1 ml de agua destilada estéril y se conservó a -20 ºC).
Posteriormente los tubos se incubaron a 56ºC durante 12 horas. Después de esto,
a cada tubo se le añadió 750 µl fenol, se mezcló en vortex
durante 30 segundos, se centrifugo a 14000 rpm por 5 min. Se extrajo 750 µl del sobrenadante y le agrego 750 µl de fenol, se mezcló en vortex durante 30 segundos, se centrifugo a 14000 rpm por 5
min. Nuevamente se extrajo 750 µl del
sobrenadante y le aplicó 750 µl de
cloroformo/alcohol isopropílico (24:1), se mezcló en vortex durante 30 segundos, se centrifugo a 14000 rpm por 5
min. Del sobrenadante se extrajo 600 µl
y se le agrego 600 µl cloroformo/alcohol isopropilico
(24:1), se mezcló en vortex durante 30 segundos, se
centrifugo a 14000 rpm por 5 min. Se extrajeron 350 µl de sobrenadante, a esto se le agrego 35 µl de cloruro de sodio 10%
del volumen (se disolvió 17.5 gr de cloruro de sodio en agua destilada y se
aforo 100 ml) y se le adiciono 700 µl
de EtOH/Amonio, 2 veces el volumen (Se disolvió 12 mg
de acetato de amonio en 100 ml de etanol absoluto y se conservó en hielo), se
mezcló en vortex durante 30 segundos, se centrifugo a
14000 rpm por 10 min. El sobrenadante se decantó y la pastilla se dejo secar en la incubadora a 37oC por 1 hora,
finalmente, se resuspendió en 30 µl de agua destilada estéril y se guardaron a –4oC para
su medición posterior.
Tratamiento
3
Técnica descrita por Penacino
(1997). Se colocaron 2 pajillas de semen bovino criopreservado
de 0.5mL o 0.25 mL en un tubo eppendorf
de 1.5 mL, a cada tubo se le agrego 400 µl de
TEC (se mezcló 100 ml de [1 ml de TRIS/ClH 1M pH: 7.5
con 2 ml de EDTA disódico 0.5M pH: 8, aforado a 100
ml con agua destilada] con 3.3 ml de cloruro de sodio 3M) ; y
20 µl Proteinasa
K (Se disolvio 10 mg de proteinasa
K liofilizada en 2 ml de agua destilada estéril), 40 µl de sulfato dodecil
de sodio (SDS) 10 % (se disolvió 50 gr de dodecil
sulfato de sodio en 450 ml de agua caliente, se ajustó el a pH 7.2 por agregado
de unas gotas de ácido clorhídrico concentrado y se aforo 500 ml) y 5 µl de DTT(se disolvió 0.154 gr. de ditiotreitol
en 1 ml de agua destilada estéril y se conservó a -20 ºC), cada tubo se mezcló en vortex
durante 30 segundos y posteriormente se incubo en baño maría durante 20 minutos
a 60 ºC. Después de esto, a cada tubo se le añadió
750 µl fenol, se mezcló en vortex durante 30
segundos, se centrifugo a 14000 rpm por 5 min. Se extrajo 750 µl del sobrenadante y le agrego 750 µl de fenol, se mezcló en vortex durante 30 segundos, se centrifugo a 14000 rpm por 5
min. Nuevamente se extrajo 750 µl del
sobrenadante y le aplicó 750 µl de
cloroformo/alcohol isopropílico (24:1), se mezcló en vortex durante 30 segundos, se centrifugo a 14000 rpm por 5
min. Del sobrenadante se extrajo 600 µl
y se le agrego 600 µl cloroformo/alcohol isopropilico
(24:1), se mezcló en vortex durante 30 segundos, se
centrifugo a 14000 rpm por 5 min. Se extrajeron 350 µl de sobrenadante, a esto se le agrego 35 µl de cloruro de sodio 10%
del volumen (se disolvió 17.5 gr de cloruro de sodio en agua destilada y se
aforo 100 ml) y se le adiciono 700 µl
de EtOH/Amonio, 2 veces el volumen (Se disolvió 12 mg
de acetato de amonio en 100 ml de etanol absoluto y se conservó en hielo), se
mezcló en vortex durante 30 segundos, se centrifugo a
14000 rpm por 10 min. El sobrenadante se decantó y la pastilla se dejó secar en
la incubadora a 37oC por 1 hora, finalmente, se resuspendió
en 30 µl de agua destilada estéril y
se guardaron a –4oC para su medición posterior. Para la
cuantificación del ADN se utilizó el método de absorción de luz ultravioleta:
Se efectuó a partir de diluciones 1/200 a 1/500 en agua destilada, para
determinar la absorción de luz UV a las longitudes de onda 230, 260 y 280 nm en un espectrofotómetro. Las relaciones 260/280 y
260/230 permitieron detectar la presencia de posibles contaminantes en las
muestras. Se calcula el contenido de ADN asumiendo que una unidad de
absorbancia a 260 nm equivale a 50 µg/ml de ADN doble cadena. Por otro
lado, se procedió a verificar la calidad de la extracción del ADN de las
muestras, en gel de agarosa al 0.8% (0.24 g de agarosa y 30 ml de buffer TBE 1X
(Tris Borato-EDTA), por medio de una electroforesis, utilizando una cámara
modelo horizon 58, (Life
Technologies®). En cada pozo se depositó un volumen final de 5 µl (2 µl de colorante Naranja G y 3 µl de
ADN), usando como buffer de corrida TBE. Las condiciones de la electroforesis
fueron de 40 minutos a 86 voltios. Transcurrido dicho tiempo los geles se
tiñeron con bromuro de etidio a una concentración final
de (1 µg/ml) durante 30 minutos. Los
resultados fueron evaluados en el fotodocumentador
(Modelo Gel Doc 2000, Marca Bio
Rad) de luz ultra violeta (UV) y se capturaron las imágenes por computadora en
el programa Quantitvone, para la evaluación de la calidad
y calidad del ADN.
A las variables evaluadas se les realizó
un análisis descriptivo (Steel, Torrie y Dickey 1997).
RESULTADOS Y DISCUSIÓN
El tiempo de ejecución para tratamiento dos fue 940 min, seguido del tratamiento uno y tres
con 825 y 100 min respectivamente. En la
Figura 1, se aprecia la calidad del ADN
extraído. En los carriles 5, 11 y 17 se
utilizaron dos pajillas de ¼ mL, en los carriles 6,
12 y 18 se utilizó una pajilla de ¼ mL, en el resto
de los carriles se utilizaron dos pajillas de ½ mL.
En los carriles 1 al 4 y 7 al 10, se aprecia la presencia de mucha proteína.
Sin embargo en los carriles donde se utilizaron una o dos pajillas de ¼ mL se observa poca proteína y mejor calidad del ADN. Del
Valle, Rodríguez y Espinoza (2004) mencionan que la diferencia en la cantidad y
calidad de ADN observada en algunas extracciones podría deberse, a la presencia
de ARN. El tratamiento tres en los carriles 13, 14, 15 y 16 no se observó ADN,
esto posiblemente debido a la cantidad de muestra utilizada, y al poco tiempo
empleado para que se llevara a cabo la lisis celular ya que este protocolo
únicamente considera 20 minutos en
incubación a 60oC. Becton Dickinson (2008) y Durviz (2008)
menciona que la baja o nula cantidad de ADN o lisis celular incompleta obtenida
de algún protocolo, en algunas ocasiones se debe al exceso de muestra
utilizado, por lo que es recomendable utilizar menor cantidad o prolongar el
tiempo de incubación en el paso correspondiente a la lisis.
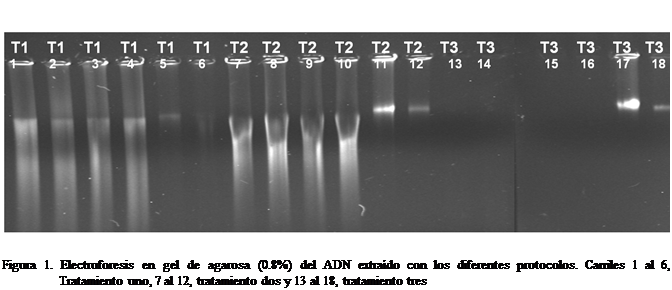
Las bandas de ADN observadas en los carriles 5, 11, 17 y 6, 12, 18,
indica una desproteinización aceptable, esto debido a
la mejor relación solución de lisis y cantidad de muestra utilizada, mientras
que el ADN observado en los carriles 1 al 4 del tratamiento uno y 7 al 11 del
tratamiento dos, presenta contaminación de proteínas, ocasionado por el exceso
de muestra. Cattaneo et al. (1997); Buttler, (2001) mencionan
que una técnica que permite extraer el ADN de un medio que contenga
contaminantes es mucho más prometedora que un método que busque extraer los contaminantes
de la preparación. El extraer el ADN con un protocolo o método adecuado
minimiza los problemas de contaminación puesto que no se requiere extraer todo
el ADN allí presente sino obtenerlo en una cantidad y calidad suficiente sobre
todo cuando éste va a ser utilizado para pruebas moleculares. La remoción de
los contaminantes del extracto es
usualmente una tarea difícil que con frecuencia lleva a inhibición de las
reacciones o errores en la amplificación (Buttler,
2001). Tratándose de métodos fenólicos de desproteinización,
la no remoción adecuada de proteínas en los extractos para la obtención de ADN
es un indicador de deficiencia en el procedimiento (Maniatis,
1982). En el Cuadro 1 se muestra la cantidad y calidad del ADN obtenida en cada
uno de los tratamientos. Para el tratamiento uno, todos los valores de la
relación A260/A280 son inferiores 0.1, lo mismo se presentó
en el tratamiento dos en los carriles del 7 al 10, esto indica presencia de
muchos contaminantes en la muestra, sin embargo, en el carril 11 se observa un
valor de 0.57, indicando este la presencia de proteínas. Para el carril 12, el
valor encontrado fue 1.45, indicando la presencia de ADN y poca contaminación
de proteínas. Para el caso del tratamiento tres, los carriles 13 al 16 no se
observaron presencia de ADN en el gel de agarosa y los valores encontrados
indican la presencia de contaminación. El carril 17 y 18, se observa un valor
de 1.28 y 2.55 respectivamente, indicando la presencia de contaminantes y de
ADN.
|
Cuadro 1. Valores de la absorbancia de ADN bicatenario
de semen bovino criopreservado extraídos con tres
protocolos. |
||
|
Carril |
Tratamiento |
Concentración (µg/mL) |
|
1 |
1 |
0.13 |
|
2 |
1 |
-0.35 |
|
3 |
1 |
-0.25 |
|
4 |
1 |
-0.48 |
|
5 |
1 |
-0.63 |
|
6 |
1 |
-0.55 |
|
7 |
2 |
-0.13 |
|
8 |
2 |
2.88 |
|
9 |
2 |
-0.40 |
|
10 |
2 |
-0.48 |
|
11 |
2 |
3.23 |
|
12 |
2 |
28.40 |
|
13 |
3 |
-0.63 |
|
14 |
3 |
-0.43 |
|
15 |
3 |
-0.43 |
|
16 |
3 |
-0.60 |
|
17 |
3 |
41.38 |
|
18 |
3 |
14.58 |
Para la relación A260 nm/A230
nm, se aprecian los valores en el Cuadro 1, el
tratamiento uno en los carriles uno al seis, siete al 11 y 13 al 16 para el
tratamiento dos y tres respectivamente, muestran la presencia de carbohidratos,
proteínas o fenoles. El carril 12 del tratamiento dos y 17 del tratamiento tres
presentan valores cercanos al rango de pureza.
Del Valle et al. (2004),
mencionan que una proporción de 0.6 para la relación (A260/A280)
corresponde a la presencia única de proteínas; y una proporción entre 1.8–2.0, corresponde
a un 90% - 100% de pureza de los ácidos nucleicos. Al respecto, Schultz et al.
(1994); Del Valle et al. (2004); De
Jesús et al. (2005) mencionan que los
valores mayores a 2.0 de la relación indican exceso de ARN en la muestra y para
la relación (A260/A230)
valores menores a 2.0 indican presencia de carbohidratos, proteínas o fenoles,
indicando también la integridad de los ácidos nucleicos (ARN), valores
inferiores a 2.0 permiten suponer la presencia de degradación de las moléculas.
En este estudio el análisis de la pureza (A260/A280) no
mostró diferencias entre los tratamientos cuando se utilizan dos pajillas de ½ mL por muestra, esto indica que la cantidad de muestra
utilizada es demasiada en relación a la cantidad de reactivo aplicado para la reacción
de lisis. El tratamiento dos mostró mejor resultado cuando se utilizó una
pajilla de ¼ mL, para el tratamiento tres el mejor
resultado se logró utilizando dos pajillas de ¼ mL.
CONCLUSIONES
Para la extracción de ADN de semen bovino criopreservado
se debe utilizar una pajilla de ¼ mL por muestra con
la técnica descrita por Penacino (1997) y dos
pajillas de ¼ mL por muestra para la técnica rápida
descrita por Penacino (1997). No se recomienda
utilizar dos o mas pajillas de ½ mL
por muestra con ningún protocolo de los aquí evaluados.
LITERATURA
CITADA
Becton Dickinson. 2008. Kit S QuickGene de extracción de ADN en Tejidos (DT-S). Para
aislamiento de ADN genómico de muestras en tejidos. Manual. Fuji Photo Film
Co., Ltd. Life science products
division . Nishiazabu
2-Chome, Minato-ku, TOKYO. JAPAN. 18 p
Boeta, M., y L. Zarco. 2000. Utilización de leche
descremada ultrapasteurizada como diluyente de semen
refrigerado de burro, destinado a la inseminación de yeguas, Veterinaria
México. Nota
de Investigación. No 001.
Buttler, J. 2001. Forensic
DNA Typing; Biology and Technology behind STR Markers. Academic, California,
USA. 322 p.
Cattaneo, C.; O. J. Craig
and N. R. Sokol. 1997. Comparison of three DNA
extraction methods on bone and blood stains up to 43 years old and
amplification of three different gene frequencies. J. Forensic Sci. 42: 1126-1135.
De Jesús, R.; N. Moreno y J. A. Martínez. 2005. Ensayo de dos
métodos de extracción de ADN de ratón para ser usado en el control genético de
ratones consanguíneos mediante la reacción en cadena de la polimerasa (PCR).
Revista Científica 15 (2): 134-140.
Del Valle, C.; A. Rodríguez y M. Espinoza. 2004.
Comparación de tres métodos de extracción de ADN a partir de restos óseos.
Complejo de Ciencias Forenses del Organismo de Investigación Judicial, Heredia,
Costa Rica. Rev. Biol. Trop. 52 (3): 717-725
Durviz, S. L. 2008. Kit extracción DNA SSS. REA1. RBME01/
RBME02. 5 pp.
Hernández, P. J. E.;
R. F. Fernández, R. Y. Gutiérrez, I. A. Córdova y N. E. Gómez. 2003. Adición de
ácido ascórbico en el diluyente para congelar semen de bovino y su efecto en la
motilidad y viabilidad de semen posdescongelado. Rev. Salud Anim. 25 (1): 39-44.
Maniatis, T.; E. F. Fritsch
and J. Sambrook. 1982. Molecular cloning: a laboratory
manual. Cold Spring Harbor Lab, New York, 468 p.
Medina Robles, V.M.; E. Sánchez Carvajal, Y. M.
Velasco Santamaria y P. E. Cruz Casallas.
2007. Crioconservación de semen bovino usando un
congelador programable (CL-8800) y determinación de su calidad postdescongelación por medio un sistema de análisis
espermático asistido por computador (CASA) Revista Orinoquia 11 (1): 75-86.
Miesfeld, R. 1999. Applied
Molecular Genetics. John Wiley, New York, USA. 293 p.
Olivares R. y R.
Urdaneta. 1985. Colección, evolución y procesamiento del semen de toros. Fonaiap Divulga (17): 4-9.
Penacino, G. A. 1997. Investigación
e implementación de sistemas de identificación de individuos por técnicas de
biología molecular, con especial referencia a los estudios post-mortem.
Tesis doctoral. Unidad de Análisis de ADN, Colegio Oficial de Farmacéuticos y
Bioquímicos, Buenos Aires, Argentina.
Schultz, D. J.; R.
Craig, D. L. Cox Foster, R. O. Mumma and J. I.
Medford. 1994. RNA isolation from recalcitrant plant tissue. Plant Mol. Biol. Rep. 12:
310-316.
Steel, R. G. D.; J. H. Torrie and D. A.
Dickey. 1997. Principles and procedures of statistics, 3rd. ed. McGraw Hill.
Yoshida, K.; K. Sekiguchi, N. Mizuno, K.
Kasai, I. Sakai, H. Sato and S. Seta, S. 1995. The modified method of
two-step differential extraction of sperm and vaginal epithelial cell DNA from
vaginal fluid mixed with semen. Forensic Science International. National
Research Institute of Police Science. Chiyoda-ku,
Tokyo. Japan p. 25-33.
Página
diseñada por Prof. Jesús Rafael Méndez Natera
TABLA DE CONTENIDO DE LA REVISTA CIENTÍFICA UDO
AGRÍCOLA


